Curium is one of the minor actinides. It has been known that its trivalent ion in aqueous solutions emits a characteristic reddish-orange fluorescence caused by the 5f electron transition. Although the experimental research team of JAERI has determined the number of hydrated water molecules, the electronic structure of the central curium ion has not been revealed, to date.
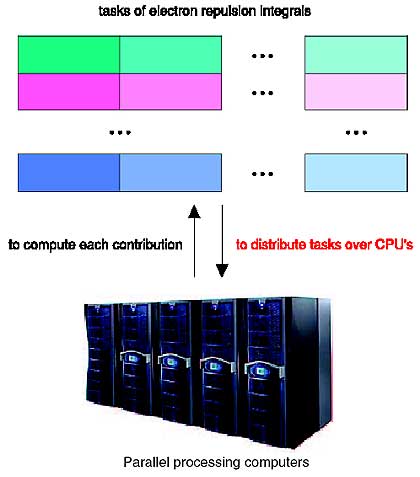
We have been promoting parallelized molecular-orbital (MO) calculations as a cutting-edge application in the high-performance computing field. In the MO scheme, the electronic wavefunction of the molecular system is expanded by pre-defined basis functions, and the expansion coefficients are determined through a sort of iterative secular equation in which a huge amount of four-indexed electron repulsion integrals are repeatedly processed until convergence. Once the wavefunction calculation has converged, the total energy, structure, property and so on are available for chemistry discussion. Although the integral processing completely dominates the total cost of MO calculations, it can be segmented to a variety of independent tasks and thus processed in a parallel fashion (Fig. 10-2). This is very promising to perform MO studies of the actinides, since the relativistic effect, which potentially demands a CPU time about one hundred times longer than that for the non-relativistic case, must be taken into account. If not parellelized, such a study is hard to perform.
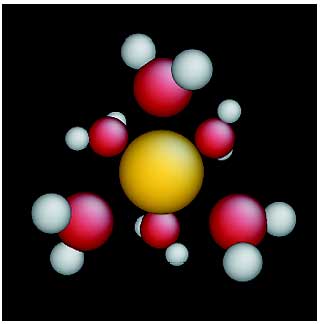
In the parallelized MO calculations with the DIRAC code, the hydrated curium ion was modeled as aqua complexes having up to six water molecules (Fig. 10-3). The following information was obtained for the first time.
(1) The electrons donated from water molecules flow mainly into the 6d shell of curium. (2) The semivalence 6p shell enhances the donation by inducing charge reorganization in the water. (3) The fluorescence transition energy of 5f is red-shifted by the presence of donated electrons.
The hydrated ion models involving other actinides can be of interest for comparison with curium. The fluorides or oxides of curium are also interesting systems, since curium in them can take the tetravalent state in contrast to gadolinium as the 4f-counter element. The parallelized MO calculations on these targets will be a future subject of study. |