 |
||
|
 |
||
|
| It is well known that the structural stability of an aggregate
composed of metal atoms, i.e. a metallic microcluster, varies
with the number of atoms in the cluster. By controlling the form
of the metallic microcluster form a few atoms up to thousands,
it is possible to design advanced materials which are different
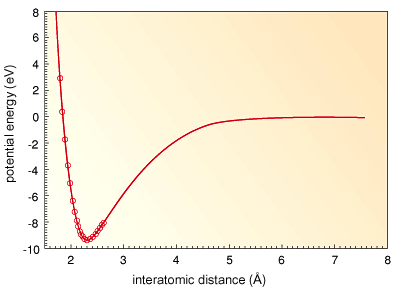
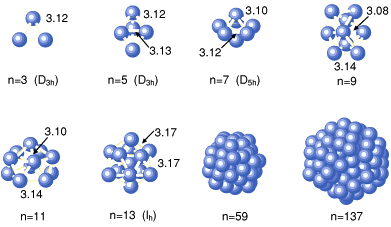
from the bulk solids. Uranium has been used as nuclear fuel, and recently, some uranium intermetallic compounds have been found to be superconducting. A lot of research has been carried out on the physical and chemical properties of these compounds. In order to understand these properties, information on the electronic structure and stability of uranium metallic clusters has become more important. We have carried out first principle electronic structure calculations for the uranium dimer and obtained the relationship between binding energy and interatomic distance (Fig. 5-7). By using the relation between the binding energy and interatomic distance, we have simulated the aggregation of uranium atoms using molecular dynamics simulations. Figure 5-8 shows predicted stable uranium microstructures. It was found that uranium clusters containing 3, 4, 5, 6, 7 and 13 atoms have highly symmetric stable structures. We also simulated the aggregation of uranium atoms to the crystal structure by increasing the number of atoms. Transuranium elements, i.e. the elements heavier than uranium, may have attractive characteristics. The information obtained from our research is likely to be very useful in basic studies of both the physical and chemical properties of these elements. |
| Reference
S. Erkoc et al., Molecular-Dynamics Simulations of Uranium Microclusters, J. Phys. Jpn., 68(2), 440 (1999). |
| Select a topic in left column |
|
Persistent Quest-Research Activities 1999 Copyright(c)Japan Atomic Energy Research Institute |