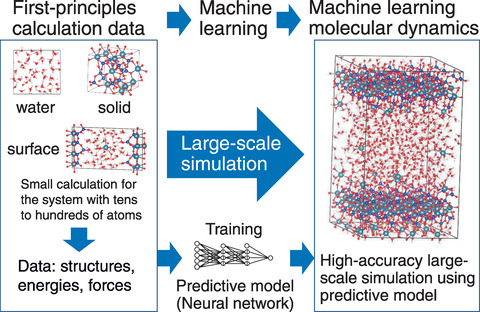
Fig.1-37 Schematic representation of machine learning molecular dynamics
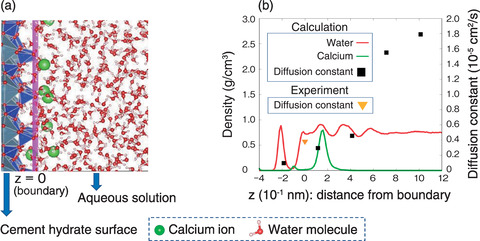
Fig.1-38 Distribution and transport properties of water and ions at the water-surface interface of cement hydrate
At TEPCO's Fukushima Daiichi NPS, due to damage to the reactor pressure vessel, a huge amount of radioactive cesium has been adsorbed into the concrete of the reactor containment vessel. For the disposal and volume reduction of large amounts of contaminated concrete obtained by decommissioning the nuclear power plants, understanding the adsorption behavior of cesium in concrete becomes important.
It is known that cesium ions are strongly adsorbed into cement hydrate (CH) in concrete, a very porous substance. Cesium ions are presumed to diffuse with water in the porous CH and adsorb onto the surface of CH. Atomic simulation is an essential tool to obtain detailed information about the form and intensity of cesium adsorption on CH. However, large-scale simulation taking into account the chemical reaction and water transport in the pores of CH is required, which is difficult to achieve with high accuracy first principles (FP) calculations due to its high computational effort.
In this study, we used machine learning molecular dynamics (MLMD) to overcome the computational cost of FP calculation. In this method, using a large amount of FP calculation data for various CH structures with tens to hundreds of atoms, a predictive model (machine learning force field: MLFF) reproducing the results of the FP calculation is created using a machine learning technique (see Fig.1-37). Molecular dynamics simulation using MLFF is called MLMD, which enables us to conduct simulation with almost FP accuracy and much lower computational effort than an FP calculation.
As a first step toward elucidating the adsorption behavior of cesium on CH, we constructed the MLFF for the basic constituent elements of CH: silicon, calcium, hydrogen, and oxygen atoms. Using the constructed MLFF, we conducted a large-scale MLMD simulation for the CH-water interface, including thousands of atoms, to investigate the transport and adsorption properties of water and calcium ions. Our simulation successfully reproduced the experimental values of the diffusion constant of water near the CH surface as shown in Fig.1-38.
In the past, accurate simulations using FP calculations have been difficult for complex systems such as CH. We have shown that MLMD enables us to perform the simulation of CH with almost FP accuracy. Future work will aim to extend the MLFF of cesium to clarify the adsorption behavior of radioactive cesium in concrete.
(Keita Kobayashi)
<Previous: 1-17 | Next: 2 Research on Nuclear Safety and Emergency Preparedness>

