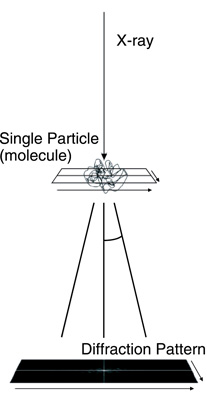
Fig.5-21 Schematic picture of single particle diffraction imaging

Fig.5-22 Electron density of a lysozyme protein and the diffraction image
Conventionally, x-ray crystallography has been the principal tool for determining the high-resolution 3D structures of proteins, nucleic acids, and their complexes. In this method, crystallization is a critical process by which a sufficiently large crystal must be prepared. However, it is known that approximately half of proteins encoded in the human genome, particularly membrane proteins, do not crystallize. X-ray free-electron lasers (XFELs), a fourth-generation light source, are extremely powerful new x-ray sources and can potentially provide a novel means for determining the three-dimensional (3D) structure of biomolecules from the diffraction images of single molecules (Fig.5-21), because coherent x-ray diffraction imaging (CXDI) using XFELs does not require a crystal. However, to realize single molecule imaging, some difficulties have to be resolved, such as the phase problem: the phase information is lost in the experimentally obtained diffraction data (Fig.5-22(b)). The phase information is needed to reconstruct the image in real space. Under certain conditions, this phase problem has been computationally solved. However, strong diffraction and prior knowledge of the size and shape of the molecule are required, but are usually unknown. This condition would be difficult to meet for single molecule CXDI, because the target of the x-rays is a single molecule, and the number of photons received by each pixel on the detector is very limited (Fig.5-22(b)), even with XFELs. Therefore, the reconstructed image with the conventional method significantly depends on the initial condition of the calculation.
Thus, we developed a new method for retrieving the phases of sparse diffraction images. The method is based on the maximum a posteriori (MAP) estimation of the Bayesian statistics and is very robust against quantum noise. Compared to the widely used conventional method, our method succeeded in reconstructing the images from diffraction data that were 10~20 times more sparse (Fig.5-22(c)(d)).
This method is a step closer to the realization of CXDI from single particles. The realization of CXDI will strongly contribute to the elucidation of the molecular mechanism of proteins and to the development of new drugs.
The present study was partly sponsored by the “x-ray Free-Electron Laser Utilization Research Project” of the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT).

