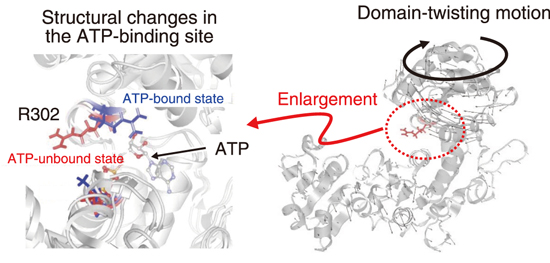
Fig.5-9 Structural changes in the adenosine triphosphate (ATP)-binding site R302 (side chain of the 302nd arginine) (Left) and domain-twisting motion (right)
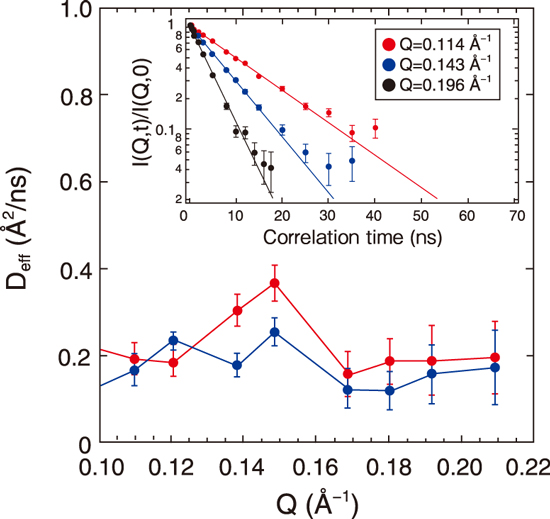
Fig.5-10 Neutron wavenumber (Q)–dependence of the effective diffusion coefficient, Deff, for ATP-unbound (red) and bound (blue) states
Proteins are composed of nanometer-sized three-dimensional (3D) structures called domains. Relatively large proteins are composed of multiple domains and are referred to as multidomain proteins. While each domain has a stable structure, the domains change their relative positions and orientations through flexible hinge regions that connect the domains. For example, the structure of the ATP-binding site changes with the domains (Fig.5-9). Elucidation of such domain-scale structural dynamics is considered necessary for understanding the mechanism of protein function expression. Neutron spin echo (NSE) technique has the highest energy resolution among quasielastic neutron scattering methods and is an effective method to capture the domain motions because it allows the observation of protein dynamics on the nanometer and nanosecond space–time scales.
Here, we focused on MurD, a typical multidomain protein. It is an enzyme involved in cell wall synthesis. Its conformational change upon binding with adenosine triphosphate (ATP) binding is essential for its function. We combined NSE with computational science to elucidate the correlation between the domain structure and structure of the ATP-binding site.
Fig.5-10 shows the NSE data, showing the effective diffusion coefficient derived from the domain motion of the protein with respect to the neutron wavenumber Q (0.1–0.22 Å-1). The peak around 0.15 Å-1 is reduced by ATP binding. Through molecular simulation, we determined the type of change in domain motion that is responsible for the reduction and found that reduction is caused by the suppression of the twisting motion of the domain (right side of Fig.5-9). Furthermore, a computational analysis showed that this domain motion is linked to the fluctuation of the side chain of the ATP-binding site (left side in Fig.5-9), indicating that the fluctuation of the local structure controlled by the domain motion is important for ATP binding. The first identification of the details of the changes in domain motions upon ATP binding has greatly advanced our understanding of the molecular mechanism of MurD enzyme activity.
These analyses reveal that the amino acid residues of the active site do not undergo random thermal fluctuations but are controlled by the cooperative fluctuations of the domain structure. The understanding the relationship of the structural dynamics between these different hierarchies is important for elucidating protein function.
This work was supported by JST-PRESTO (JPMJPR14L7).
(Hiroshi Nakagawa)

