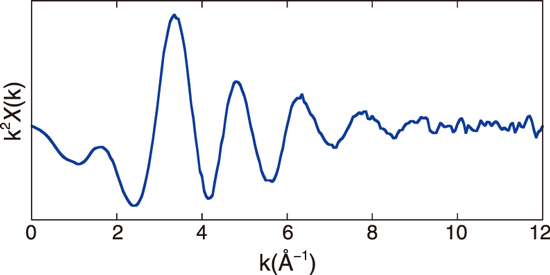
Fig.9-7 Extended X-ray absorption fine structure (EXAFS) spectrum of the hydrated Ba2+
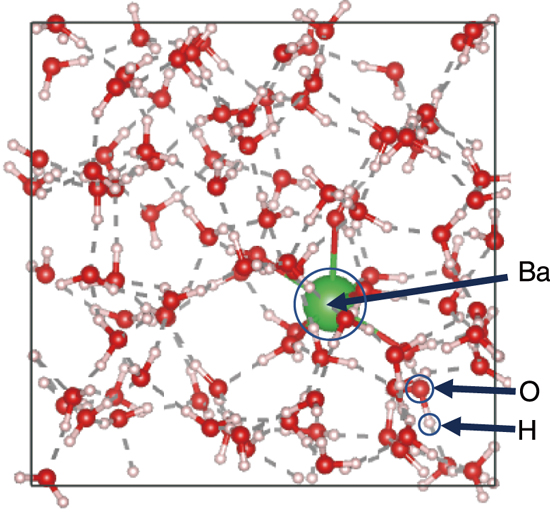
Fig.9-8 Snap shot of the ab initio molecular dynamics (AIMD) simulation
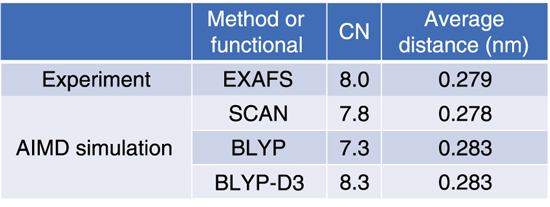
Table 9-1 Obtained coordination numbers (CNs) and average distances
Radium (Ra) is an important element to be considered with regard to the disposal of radioactive waste and environmental problems related to uranium (U) ores because Ra is formed by the decay of U and thorium (Th). However, the fundamental properties of Ra, such as hydration structure, remained unclear because of the difficulty in conducting experiments on Ra. In this study, barium (Ba), an analog element of Ra was examined. Ba is an alkaline earth metal element, similar to Ra, and its ionic radius is close to that of Ra2+. Because of these similarities, Ra and Ba have similar chemical properties. The purpose of this study is to investigate the hydration structure of Ba2+ via extended X-ray absorption fine structure (EXAFS) and ab initio molecular dynamics (AIMD) analyses.
EXAFS is a powerful tool to directly reveal the local structure of the target element. The strengths of EXAFS are its high applicability and element selectivity. This study conducted EXAFS measurement for the Ba K-edge at beamline NW10A in Photon Factory Advanced Ring (PF-AR). The measured sample was a 0.5-M solution of barium nitrate, stored in a plastic bag. The obtained spectrum is shown in Fig.9-7.
For AIMD simulations, the exchange-correlation (XC) functional is essential. Previous studies showed that the recently developed strongly constrained and appropriately normed (SCAN) functional is appropriate for simulating the hydration structures of monovalent ions. Therefore, we considered that the SCAN functional suitable also for divalent ions such as Ba2+. In this study, the commonly used Becke–Lee–Yang–Parr (BLYP) and BLYP with dispersion effect (BLYP-D3) functionals were used for comparison in addition to the SCAN functional (Fig.9-8).
The EXAFS and AIMD studies independently revealed the hydration structure of Ba2+, the number of water molecules in the first hydration shell (i.e., coordination number; CN), and the average distance between Ba2+ and its neighboring oxygen atoms (Table 9-1). Therefore, the values obtained by AIMD simulation with the SCAN functional were the closest to those obtained by EXAFS. These results imply that the SCAN functional is appropriate for simulating the hydration structures of divalent ions, and hence, it is expected to be applicable for other divalent ions such as Ra2+. As future work, the methods used in this study will be applied for examining the hydration structure of Ra2+ and quantitative comparison between Ba2+ and Ra2+.
This study was conducted in collaboration with the University of Tokyo and Osaka University and supported by JSPS KAKENHI Grant-in-Aid for Research Activity Start-up (JP19K23432).
(Akiko Yamaguchi)
<Previous: 9-3 | Next: 10 Development of Science & Technology for Nuclear Nonproliferation>

