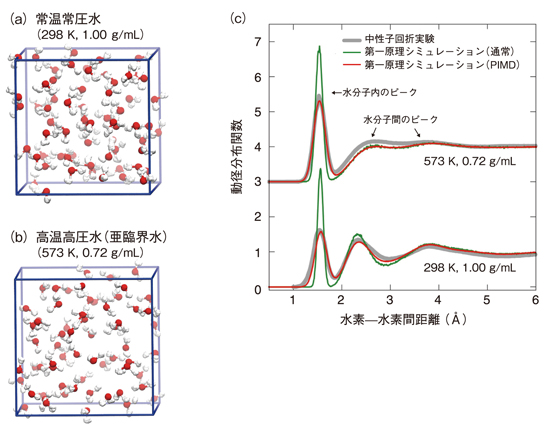
図9-6 水の構造と原子間動径分布関数
物質は原子で構成されており、原子の集団運動を追随する分子シミュレーションを行うことで、物質の性質を知ることできます。原子運動は各原子が持っている電子の振る舞いによって決まるため、電子の振る舞いを支配している量子力学を用いて計算する「第一原理計算」と呼ばれる手法によって電子の振る舞いを明らかにし、その結果に基づいてさらに原子運動を計算する「第一原理シミュレーション」が必要となります。第一原理シミュレーションは、高性能の大型計算機を利用することで、これまで知られていなかった物質の微視的振る舞いを続々と解明してきています。そうした中で、私たちの最も身近な物質の一つといえる水の構造については、従来の第一原理シミュレーションでは実験結果を再現できないことが知られていました。それは、水分子に含まれている水素原子が非常に軽いため、電子と同じように量子力学の影響を受けるので、通常の第一原理シミュレーションでは考慮されていない「量子ゆらぎ」(物体の位置が確率的にしか存在できない状態)を示すためです。特に水分子から他の分子へ水素が移るような化学反応では量子ゆらぎの影響が大きいため、この影響を第一原理シミュレーションで正確に計算できれば、原子燃料の再処理、環境中の放射線元素の動態などを含む、種々の水中での化学反応をより正確に評価することができます。それには、電子の第一原理計算に加え、原子核の量子ゆらぎを扱った計算が必要です。そこで、システム計算科学センターでは、原子力分野の基盤研究技術として、原子核の量子ゆらぎを取り入れた計算手法とそのソフトウェア「PIMD」を開発してきました。これを用いて、水の第一原理シミュレーションを行いました。図9-6では、動径分布関数(原子間距離の分布)について、通常の第一原理シミュレーションと、PIMDを用いて量子ゆらぎを含めた第一原理シミュレーションを中性子回折実験(結晶による中性子の回折現象を利用し物質の構造を解析する実験)と比較しています。水素原子の量子ゆらぎが大きいほど、グラフのピークが低く幅が広くなります。通常の第一原理シミュレーションでは実験と乖離する部分があるのに対し、PIMDを用いたものは実験とよく一致しており、水分子から他の分子へ水素が移るような化学反応をより正確に評価可能となることが期待できます。
PIMDはシステム計算科学センターのウェブサイト(https://ccse.jaea.go.jp/software/PIMD/index.jp.html)でオープンソースソフトウェアとして公開され、国内外の研究者によって利用されています。PIMDの特徴は、様々な第一原理計算コードと組み合わせて、原子核の量子ゆらぎを取り入れた計算などの高度な第一原理シミュレーションが可能なことであり、物理学、化学、材料科学分野の研究で役立っています。
(志賀 基之)

