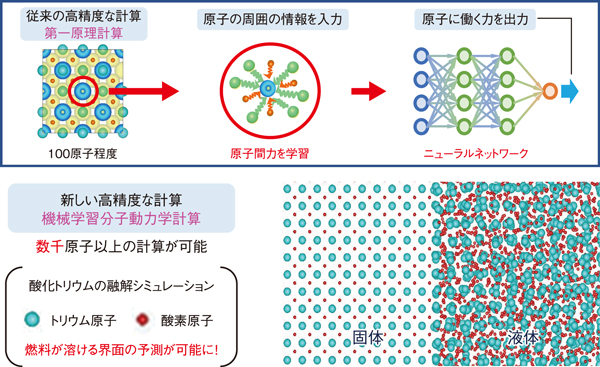
図1 機械学習分子動力学法の概略図
原子力の利用やその研究開発において、核燃料の安全性を担保し、核燃料が発するエネルギーを効率良く利用するためには、核燃料物質の比熱の温度依存性や融点等の高温での物性を正しく理解することが必須となります。しかし、核燃料の物性を調べるための実験は難しく、特に高温では詳細にデータを集めることは困難でした。そのため、希少な実験データを補うため、原子レベルの高精度シミュレーションに対し大きな役割が期待されています。
しかし、従来の原子レベルのシミュレーションでは、高精度な第一原理計算を用いると計算コストが膨大になる一方で、計算コストの低い分子動力学計算を採用すると、精度が下がり、実験結果の再現が困難になるというジレンマに陥っていました。本研究では、高精度な第一原理計算結果を機械学習し、低計算コストで高精度な分子動力学計算を実現可能な機械学習分子動力学法と呼ばれる手法を用いて、この問題の解決に取り組みました。
具体的な対象として、次世代の核燃料物質として注目を集めている、酸化トリウムの高温物性の機械学習分子動力学計算を実施しました。機械学習分子動力学法では、数千通りの異なる原子配置に対する第一原理計算を行い、ニューラルネットワークにより分子動力学計算で必要となる原子間の力を学習します(図1)。このニューラルネットワークを利用した機械学習分子動力学計算により、酸化トリウムの高温物性の評価に必要な大規模計算を低計算コストで実施することに成功し、高温での比熱、融点等の高温物性を高精度に再現することに成功しました。
機械学習分子動力学法により大規模かつ高精度な分子動力学計算が可能になったことで、核燃料物質の詳細な物性を原子レベルのシミュレーションにより取得可能としました。この手法により、通常運転時のみならず、シビアアクシデント時のように、実験困難な超高温の核燃料の状態を推定し評価することが可能となります。本開発手法は、トリウム燃料だけでなく、ウラン燃料やMOX燃料への適用も可能であり、今後、それらの物質についても同様の研究開発を進める予定です。
本研究の一部は、日本学術振興会科学研究費基盤研究(C)(18K05208)「機械学習分子シミュレーションと実験による粘土鉱物界面水物性の解明」、基盤研究(C)(18K11345)「エクサスケール計算機を想定した量子モデルシミュレーションに対する並列化・高速化」の助成を受けたものです。
(小林 恵太)

